Troubleshooting
Listed in this article are some of the common pitfalls, errors, and warnings that the user may encounter when using cytomarker. We have attempted as much as possible to explain some of these common alerts so that the user may continue with his/her analysis.
Common Errors
Errors are messages in the run that will stop the current analysis and prevent any further analysis from occurring. Below is a list of some common error messages that the user may encounter:
No cells remain after filtering/subsetting
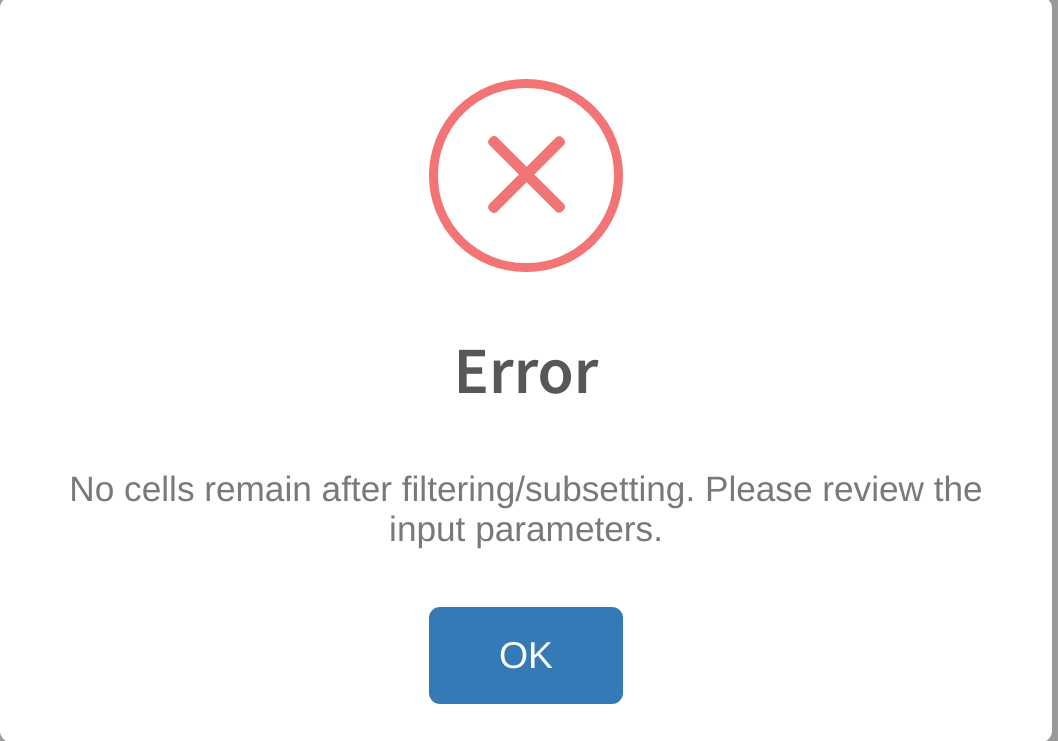
This error may occur if the user sets the filtering thresholds too high (i.e. by selecting a minimum cell category cutoff that is greater than the largest grouping in the Cell category to evaluate selection). Review the thresholds set for the cell category under Advanced Settings -> Category subsetting and adjust tjhe threshold accordingly to ensure that at least 2 groupings from the cell category are included in the analysis.
Error: more than 100 unique elements
An error message similar to the one below;
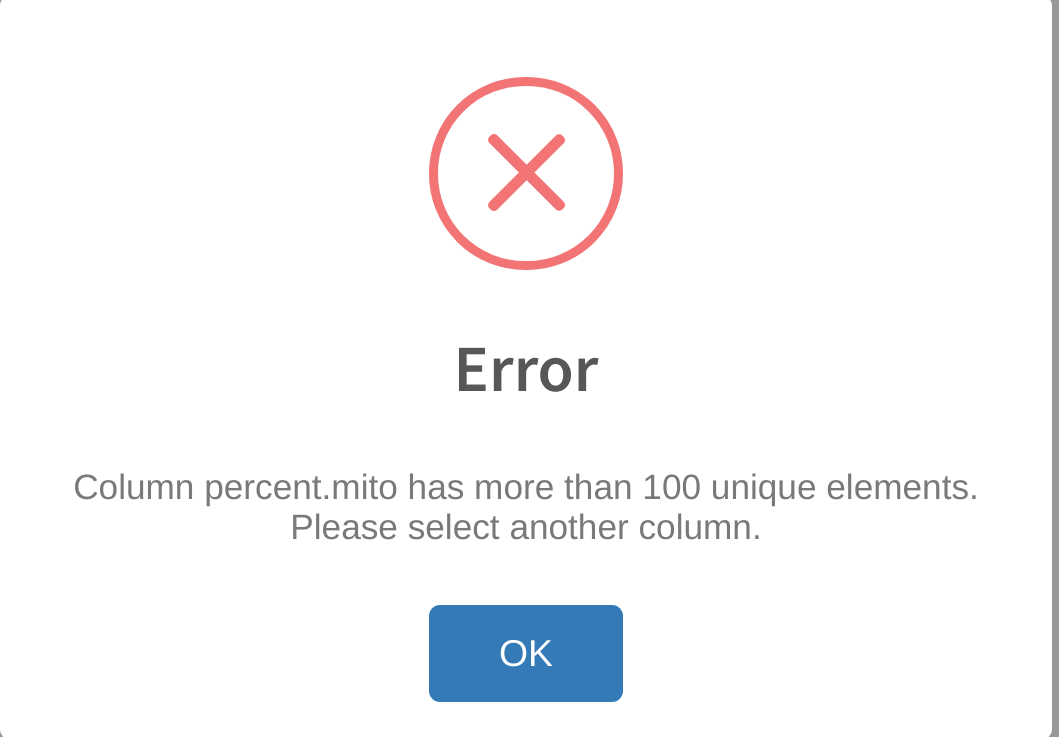
indicates that the user attempted to select a cell category in Cell category to evaluate that has too many unique levels (greater than 100) for analysis. cytomarker limits the number of unique factors in a cell category in order to
- provide more accurate scoring to the provided cell types, and
- to be able to render the plots in a comprehensive and visually appealing way
Cell categories with over 100 unique types compromise the interpretability and visual quality of the analysis. The user should select a different cell type category of interest that has fewer than 100 levels. Suggested cell type categories include cell type annotations, time points, patient/disease states, different experimental conditions, etc. Users may also choose to view a curated dataset using Select a curated dataset under Get Started.
Error: invalid genes in the current panel
An error message similar to the one below:
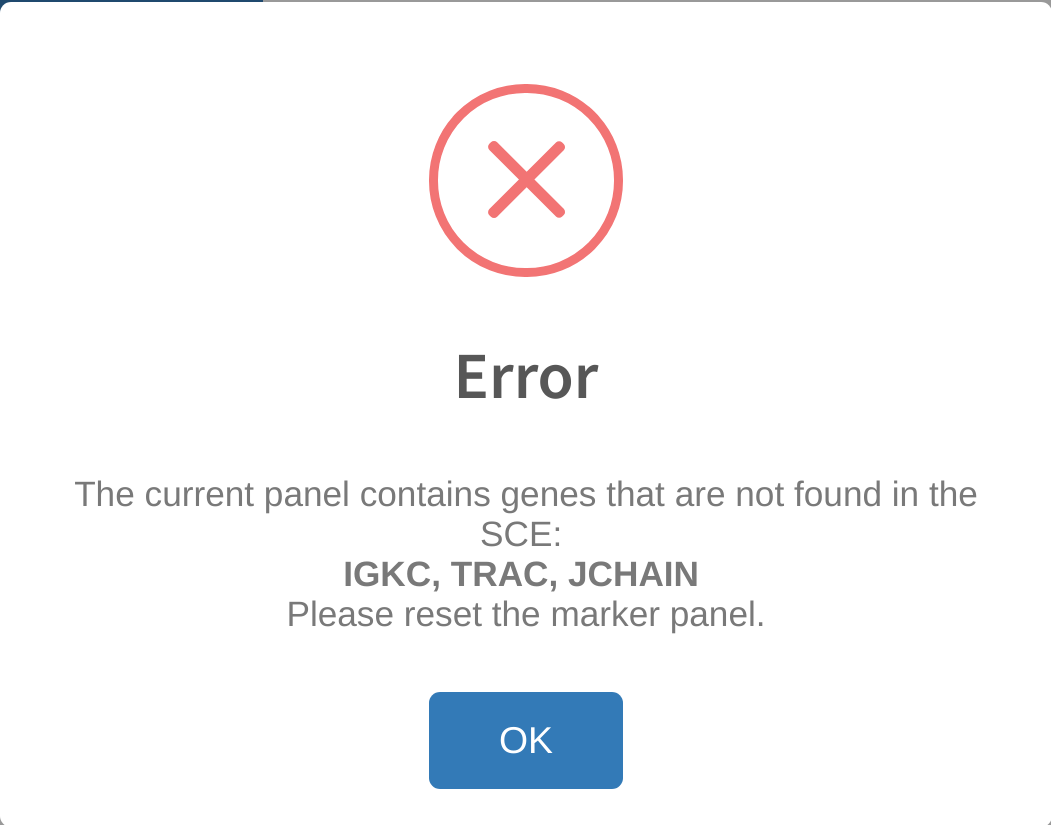
indicates that the user is attempting to use a current panel with top/selected markers that cannot be found in the gene names for the loaded dataset. This can occur if a) The user has previously generated the existing panel using a different dataset, then loaded a new one into memory, or b) if the user has loaded a panel from a previous analysis using a saved yml file. In either instance, the analysis cannot proceed because the markers have no count measurements in the dataset. The user can either load a different dataset that contains these genes, or reset the marker panel and re-generate a set of markers that are relevant to the genes in the dataset.
Common Warnings
Warnings are messages that provide an alert or notification to the user about some unique aspect of the analysis that should be noted. These messages do not prevent any further analysis from occurring. It is important that the user be familiar with some of the common warnings messages as these often affect the final run outputs and interpretability of the results.
Warning: genes that are markers for multiple cell types

This occurs when cytomarker detects that there is redundancy in the marker panel being generated; that is, it detects that a particular gene may be an appropriate marker for multiple cell types in the Cell category to evaluate selection. This is likely to occur if:
- The user sets the desired panel size to be a very large value i.e. 100 or greater
- The user uses a very small dataset, with either very few genes or litle count information
In either case, cytomarker will likely not generate a marker panel list of the desired size, as redundant genes will be removed to improve the cell type scoring. The user should review the desired panel size and quality of the count information to determine if there is sufficient information to infer distinct cell types, and/or if the desired panel size is too large. cytomarker sets a default panel size of 32, and the suggested upper limit of the panel size should not exceed 50-60 in most analyses.
Warning: cell types with abundance below set threshold
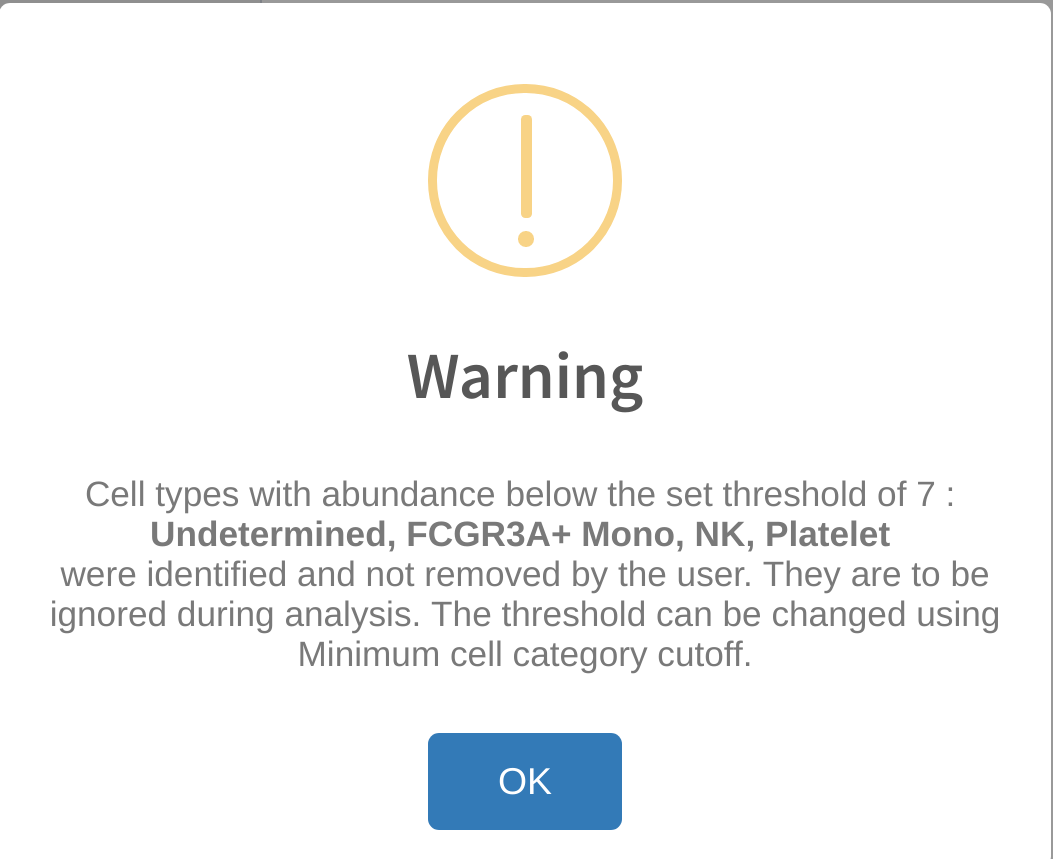
By default, cytomarker requires a minimum of 2 cells per cell type level in the Cell category to evaluate selection in order to perform scoring. Therefore, any cell type categories that have fewer than 2 cells are automatically ignored (i.e. a user performing immune cell profiling with just one NK cell in his/her dataset will not receive any results for NK cells.) This threshold can be modified in the Category subsetting field of Advanced Options to increase the threshold for cells to ignore. Is is suggested to ignore cell types with an abundance that is very small in proportion to the overall size of the dataset. For example, a dataset with 60,000 cells which contains just 5 glial cells, while still meeting the minimum threshold, is not likely to perform well in the machine-based scoring analysis. The user should therefore consider what minimum threshold to set for cell type levels in order to achieve good scoring on all cell types in the output. The count and proportion distribution of the cell types can be reviewed in ``Category subsetting` prior to setting the thresholds.
Any cell types below the set threshold will be ignored during analysis and shown in a warning message to the user. If desired, the user can later go back to the original dataset and re-add these cell types to compare the performance of different cell types across runs.
Reupload warnings: incompatible dimensions or cell type category not found
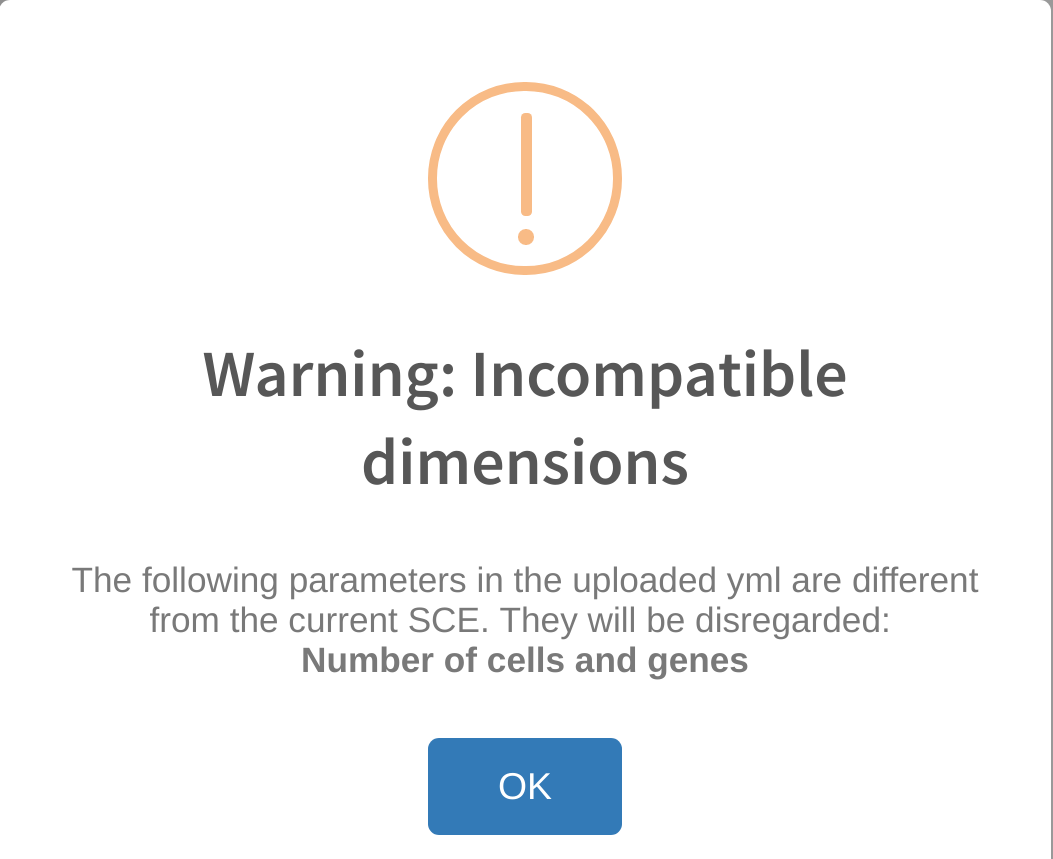
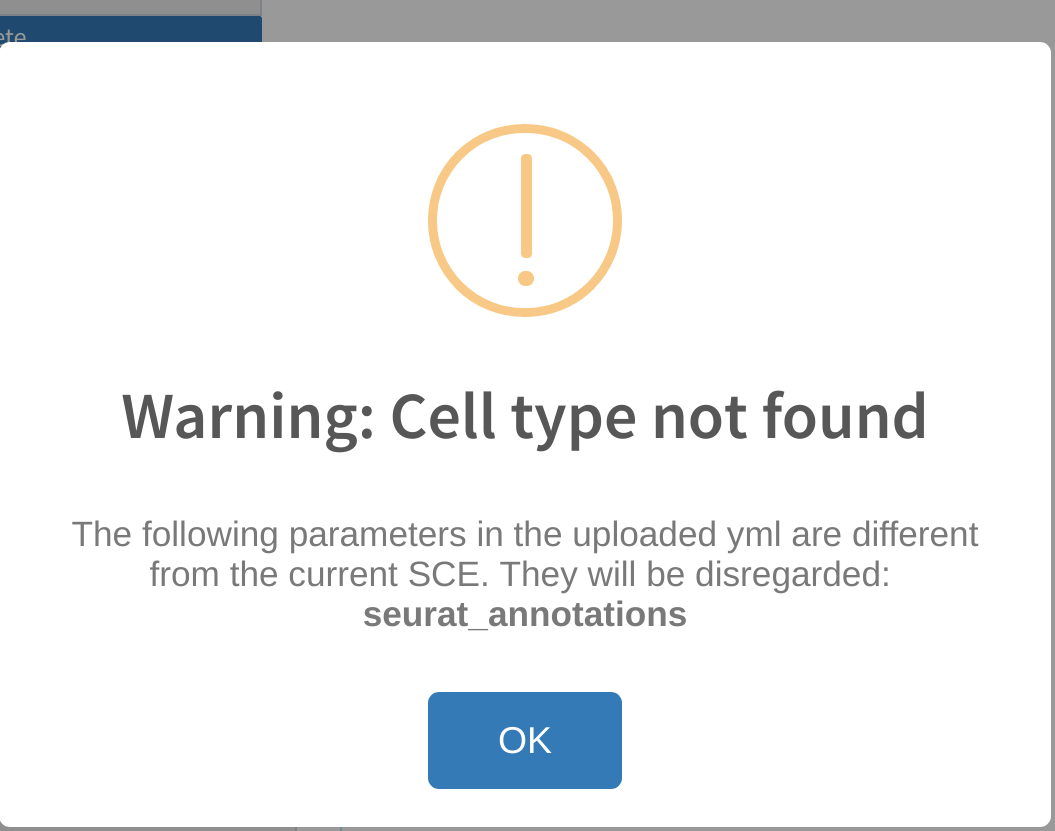
Errors similar to the ones above are likely to occur if the user attempts to upload a previous analysis yml file that was generated with a different dataset. When cytomarker exports the run parameters into a yml file, it includes the dimensions of the dataset used to generate the analysis. While cytomarker accepts using previous run yml files on different datasets during reupload, it does pose some possibile problems, such as the marker panel containing genes that are not found in the current dataset.
cytomarker will inform the user of any differences in compatible parameters between the reuploaded yml file and the current dataset. In some instances, the user may need to manually set certain run parameters if they have not been automatically set from the reupload, such as Cell category to evaluate
App crashes
When cytomarker crashes, there will likely be no warnings or alerts provided to the user. In this case, the user should be prompted to review the following elements of the analysis, as certain deviations from desired inputs may cause the analysis to crash without warning. Note that the developers of cytomarker have tested the app extensively on canonical scRNA-seq inputs but cannot always anticipate the performance when running any analysis on edge cases, low-quality or very large datasets.
-
Cell type proportions: the user should review the cell type proportions of *
Cell category to evaluateunderAdvanced settings -> Category subsetting. Cell type factors that are less than 1-2% of the total dataset population may cause problems during subsetting, as there is a possibility that only one cell from this level is selected at random for analysis (cytomarker requires a minimum of two cells per cell type factor for scoring). The user should exclude these lowly expression cell types from analysis. -
Check overall dataset size and quality: Datasets with fewer than 100-200 cells may not perform properly during analysis as the count information may be too sparse to run the ML scoring model. Additionally, cytomarker may struggle to find suitable marker genes for certain cell types if the count information is too sparse. Conversely, datasets that are too large (several 10,000s to 100,000s of cells) may take too long to upload to cytomarker on a poor internet connection. The user should aim to retain between 2000-10,000 cells for analysis, and make use of the subsetting feature under
Advanced settingsif the number of cells uploaded exceeds 2000.
App is frozen when computing the UMAP coordinates
Occasionally, the application will run out of memory when attempting to compute and render the UMAP plot. In this instance, it is suggested to refresh the application, and ensure that Precomputed UMAP under the Advanced Settings is selected. If this option does not exist, the user is strongly recommended to create precomputed UMAP coordinates for the specific dataset locally, which can be used each time cytomarker performs an analysis run. Visit the data preparation guide for guidelines on how to compute the UMAP for an scRNA-seq dataset.